





-
不同于传统的 X、
$\gamma $ 等射线,重离子辐射具有独特的物理和生物学特征,主要包括线传能密度(Linear Energy Transfer, LET)高,能量沉积的射程分布为Bragg曲线等[1]。重离子在击中DNA的靶点区域时通常诱发复杂的团簇型损伤,即在有限的空间内产生多位点、多类型的DNA损伤,进而诱发显著的生物学效应[2]。重离子辐射诱导的DNA团簇损伤包括多种损伤类型,因此其修复也涉及到不同的修复机制,当多种修复蛋白聚集在一个有限的空间时,修复过程更容易产生“错误”,进而产生更为复杂的突变模式[3]。辐射诱变发生在DNA水平,但目前的实验方法,如免疫荧光技术、DNA损伤修复报告基因等只能确定重离子辐射靶点的空间位置,尚不能在序列水平对重离子击中DNA的靶点进行有效的定位,这也是相对于 X 和$\gamma $ 辐射,重离子辐射诱变机制研究较为滞后的一个重要原因。重离子击中DNA靶点时通常会产生各种类型的DNA损伤,然而在团簇的中心区域富含DNA双链断裂(Double-Strand Break, DSB)[4]。DSB是辐射诱导生物学效应中最重要的原发事件,未修复的DSB通常会引起遗传信息的丢失甚至导致细胞死亡[5]。针对DSB损伤的修复通路主要有非末端同源连接(Non-Homologous End Joining, NHEJ)和同源重组(Homologous Recombination, HR)两种。NHEJ能够对DSB进行快速、有效的修复,但NHEJ不需要同源DNA作为模板,因此在修复过程中更容易引入突变。相对而言,HR利用未受干扰同源的DNA链作为模板来修复受损的DNA序列,尽管修复效率较低,但修复精确度很高[6]。HR又分为保守的SDSA(Synthesis-Dependent Strand Annealing)途径和非保守SSA(Single-Strand Annealing)途径,其中前者被认为是HR的经典通路[7]。由于基于HR的报告基因构建方法和原理相对成熟,且有多个重组报告体系被报道,这些体系主要被用来评价各种胁迫因子诱导的DSB程度[8-13]。考虑到重离子团簇损伤的中心区域富含DSB的特性[14],本研究提出用同源重组机制对重离子的辐射靶点进行序列定位,并通过检测重组元件侧翼序列来分析重离子辐射靶点诱变特征的研究思路。
基于上述研究思路,本研究主要利用大肠杆菌生长周期短,突变检测方法成熟等优势,构建了TetA同源重组元件用于重离子辐射诱导团簇损伤的序列定位,并在TetA重组元件侧翼连接突变筛选基因LacI高效检测DNA团簇损伤演化的遗传突变。在体系构建的基础上,利用
$\gamma $ 和碳离子进行辐射处理,检测TetA 元件重组特性及侧翼LacI基因的突变情况,初步证明上述研究思路的可行性,为进一步研究重离子辐射诱变的相关机制提供了方法学基础。 -
$\gamma $ 辐照处理采用BIOBEAM GM2000型$\gamma $ 射线辐照仪(Biobeam GM2000, Leipzig,德国),放射源是137Cs密封源,剂量率2.5 Gy/min。碳离子辐照(80 MeV/u,剂量率20~50 Gy/min,LET为30 keV/µm)依托中国科学院近代物理研究所重离子加速器平台(HIRFL)进行。 -
实验所用的LacI基因缺失型大肠杆菌(LacI-)以野生型大肠杆菌MG1655(中国科学院天津工业生物技术研究所惠赠)为背景,利用CRISPR-cas9技术敲除基因组LacI基因构建而成。克隆载体PEASY-Blunt Cloning Kit购于北京全式金生物技术股份有限公司。LacI-大肠杆菌感受态细胞制备参照Inoue“超级感受态”细胞制备的方法[15]:LacI-感受态细胞菌株在LB固体培养基Z字形划线,置于37 ºC恒温培养箱培养12 h,挑取单克隆到SOB液体培养基中,18 ºC,180 r/min恒温摇床培养16 h,测出大肠杆菌OD600为0.4~0.6,使用低温冷冻离心机4 ºC 2 500 g离心10 min,弃上清。加入适当比例的Inoue转化缓冲液,4 ºC 2 500 g离心10 min,加入适当比例的Inoue缓冲液以及DMSO,混匀并进行分装,制备好的大肠杆菌感受态细胞于−80 ºC保存。
-
使用融合聚合酶链式反应(Polymerase Chain Reaction, PCR)技术,设计具有互补末端的引物(表1),形成具有重叠链的PCR产物,再通过PCR产物重叠链部分的延伸,在不需要内切酶消化以及连接酶处理的条件下实现不同来源的DNA片段在体外的连接。首先构建含有同源序列的TetA基因片段,随后将构建成功的含有同源片段的TetA基因与LacI基因片段进行连接。连接成功的片段经PCR验证,并经过测序证明序列无误后进行下步实验。
表 1 融合PCR引物列表
引物名称 引物序列 TetA-2F GCTTGACACTTTATCACTGATAAAC TetA-R TCAGCGATCGGCTCGTTG LacI-F ACACCATCGAATGGCGCAAAACC LacI-R TCACTGCCCGCTTTCCAGTCGG LacIF-TetR-R GGTTTTGCGCCATTCGATGGTGTTCAGCGATCGGCTCGTTG TetR-LacIF-F CAACGAGCCGATCGCTGAACACCATCGAATGGCGCAAAACC -
取连接好的TetA-LacI基因片段5 µL与克隆载体PEASY-Blunt Cloning Kit 1µL在25 ºC的温度下进行平末端连接,将含有连接产物的质粒转化进入LacI- 大肠杆菌感受态细胞,5 µL的质粒转化LacI- 100 µL的感受态细胞,冰浴30 min,随后42 ºC热激2 min,热激过后继续冰浴2 min,加入900 µL的液体LB培养基将菌液补足至1 mL,37 ºC,200 r/min,培养1 h。PEASY-Blunt Cloning Kit克隆载体自身带有卡那霉素抗性,将培养好的菌液涂至含有卡那霉素抗性的LB固体培养基中,37 ºC培养12 h,含有TetA-LacI基因片段的质粒的转化子PEASY-Blunt-Tet-LacI可以通过卡那霉素抗性筛选获得。为了排除筛选到的单克隆为假阳性的情况,使用克隆载体PEASY-Blunt Cloning Kit的通用引物 M13F&M13R,将在卡那霉素抗性板上筛选得到的单克隆菌落作为模板DNA 进行 PCR 扩增并验证产物长度。
-
小剂量质粒抽提试剂盒购于上海生工生物股份有限公司。
将含有TetA-LacI基因片段的质粒的转化子PEASY-Blunt-Tet-LacI单克隆挑至10 mL含有卡那霉素抗性的液体LB培养基中,37 ºC,200 r/min培养,酶标仪检测到菌液OD600为0.5左右时,将菌液进行分装,分别进行
$\gamma $ 100 Gy、重离子100 Gy 的辐照。由于质粒在细胞中是高拷贝,难以同时分析发生在同一个质粒上的TetA基因重组和LacI基因突变,所以辐照处理后需将质粒提取并再次进行转化,确保重组和突变的质粒能够被特异性筛选。 -
不同剂量辐照后的菌液进行提质粒并重转化后,将含有转化子PEASY-Blunt-Tet-LacI的菌均匀地涂在添加四环素和卡那霉素抗性(统计四环素重组细胞)以及单独含有卡那霉素抗性的固体LB培养基(统计总的细胞数)上。未发生同源重组修复的质粒,四环素抗性基因没有活性,不能被四环素抗性板筛选。37 ºC培养16 h后,对培养板上的单克隆进行统计。四环素重组率(10n-m)=TetA基因重组细胞数(10n)/总细胞数(10m)。
-
提取不同剂量辐照后菌的质粒并重转化后,将含有转化子PEASY-Blunt-Tet-LacI的菌均匀地涂在含有卡那霉素抗性的P-Gal培养板(统计LacI基因突变的细胞)和单独含有卡那霉素的固体LB培养板(统计总的细胞数)上。37 ºC培养72 h后,LacI基因发生突变,LacZ产生半乳糖苷酶并分解苯基-β-D-吡喃半乳糖苷(P-Gal)产生半乳糖以供大肠杆菌利用,长出正常菌落,而LacI未发生突变的大肠杆菌由于半乳糖苷酶的表达量极少,72 h内不会产生可见菌落。因此用P-Gal作为唯一糖源的基础培养板可以用来筛选LacI突变子。将P-Gal+Kan抗性板上筛选得到的的单克隆重新挑菌至含有卡那霉素抗性的液体LB培养基中,待菌液浑浊后,将菌液点涂至含有X-Gal的Kan+LB培养基上,进行蓝白斑筛选复筛,LacI基因突变可以使LacZ基因正常产生β-半乳糖苷酶,从而将无色化合物X-Gal切割成半乳糖以及深蓝色的5-溴-4-靛蓝。对培养板上的蓝色单克隆菌落进行统计,并测序验证。LacI突变率(10n-m)= LacI基因突变细胞数(10n)/总细胞数(10m)。
-
100 Gy
$\gamma $ 和碳离子辐射处理后,提取菌液质粒并再次转化后,将含有转化子PEASY-Blunt-Tet-LacI的菌液均匀地涂在含有卡那霉素抗性的P-Gal培养板上,并统计LacI基因突变的细胞数(P-Gal培养板为营养缺失型培养板,无法同时加入卡那霉素与四环素两种抗生素),同时接种含有卡那霉素的固体LB培养板统计总的细胞数。37 ºC培养72 h后,将P-Gal+Kan抗性板上筛选得到的单克隆重新挑菌至含有卡那霉素抗性的液体LB培养基中,待菌液浑浊后,将其接种在含有X-Gal的LB+Kan+Tet固体培养基中,如果菌落在含有Kan+Tet双抗性的LB培养基中可以正常生长,并且菌落呈现蓝色,表明筛选到了TetA基因发生同源重组及LacI基因产生突变的双突变克隆,并进行Sanger测序验证,筛选得到阳性双突变克隆,突变频率计算公式为:(Tet基因重组+LacI突变率)(10n-m)=(TetA+LacI突变细胞数)(10n)/总细胞数(10m)。 -
每个辐照实验重复3次,每次试验设置3个平行组,试验结果统计后算得平均值。实验数据的组间差异分析均采用单因素方差分析。P < 0.05为差异有统计学意义,P < 0.01为统计具有显著性差异。统计和绘图均使用Graphpad 8软件完成。
-
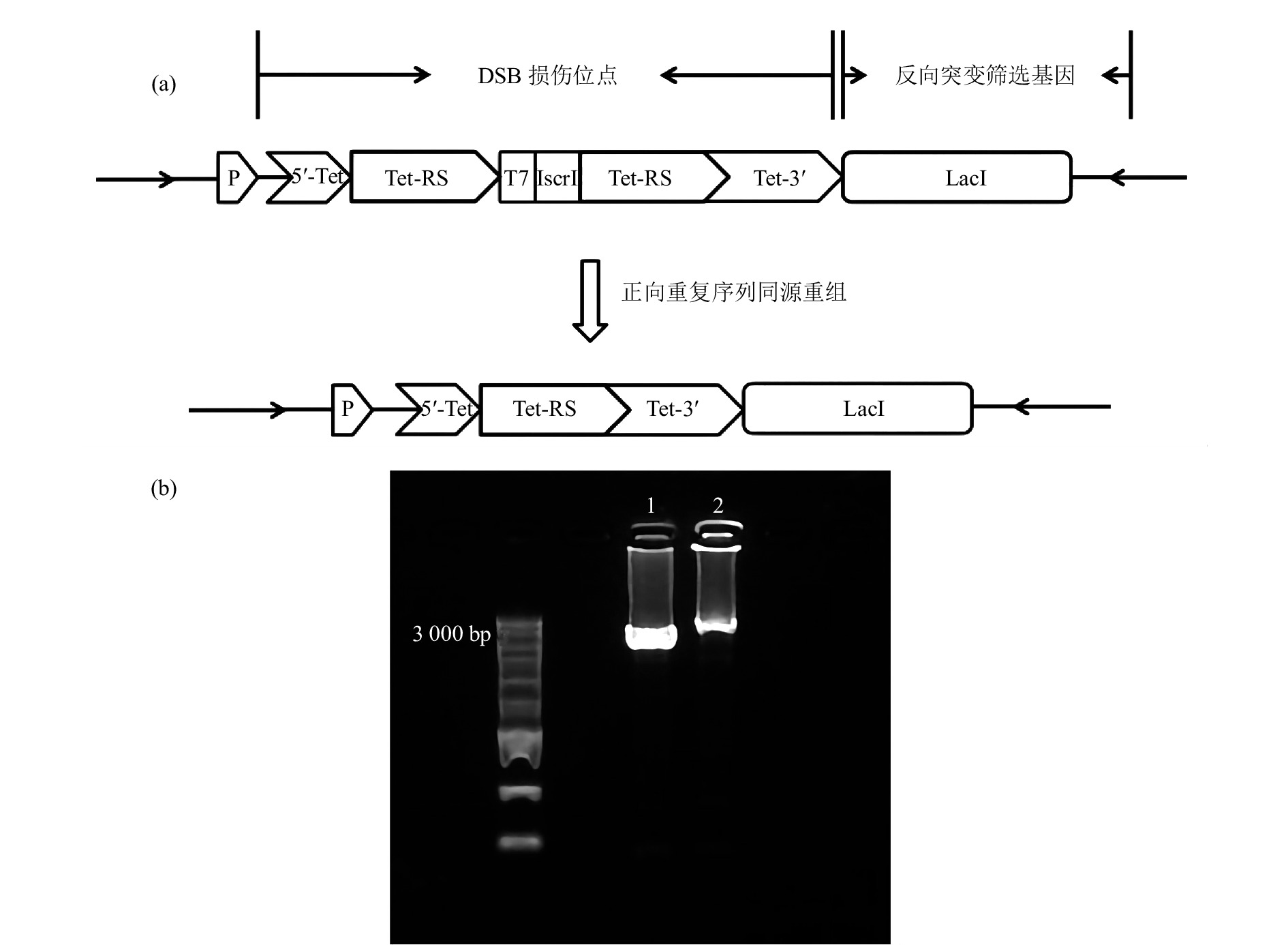
为了研究重离子诱导的团簇损伤引发的突变特征,在PEASY-Blunt Cloning Kit质粒上构建了基于TetA同源重组元件的重离子辐射DNA损伤位点标记及突变分析体系[图1(a)]。以全长1 200 bp的TetA基因为同源重组标记DSB损伤位点。TetA基因的启动子在TetA起始密码子的前200 bp处,5’Tet设计片段为从起始密码子ATG 开始到第61 bp,随后TetA基因碱基序列从61bp处连接上相同的两段同源重复序列Tet-RS,Tet-RS分别设计有300 bp(同源序列为TetA基因的61-361 bp)、600 bp(同源序列为TetA基因的61~661 bp)、900 bp(同源序列为TetA基因的61~961 bp)三个长度,并在两段同源重复序列中间加上T7终止子以及一个IsceI内切酶位点(用于是否发生重组的双重验证,发生重组会导致IsceI内切酶位点删除)。5’Tet+Tet-RS有启动子活性,但是不具有TetA基因的完整序列,而Tet-RS+Tet3’因为中间连接有T7终止子的原因,不具有TetA基因的启动子活性,这种结构导致TetA基因不具有活性(TetA−)。辐射产生DSB的位点是偶发性的,但是当DSB损伤位点产生于两个同源重复片段Tet-RS的任何一个序列上(只有DSB可以导致DNA同源重组),通过序列重组删除一个同源重复片段Tet-RS以及中间序列,即可恢复TetA基因活性(TetA+)。利用TetA同源重组报告基因可以特异性标记发生DSB的位点,进一步在其侧翼序列直接连接反向突变筛选基因LacI,LacI基因全长1 160 bp,两段基因序列中间无space片段,用于分析DSB位点附近的突变情况及突变特征。并将该片段插入到PEASY-Blunt Cloning Kit克隆载体的多克隆位点,并将质粒转化到LacI缺失型大肠杆菌感受态细胞。
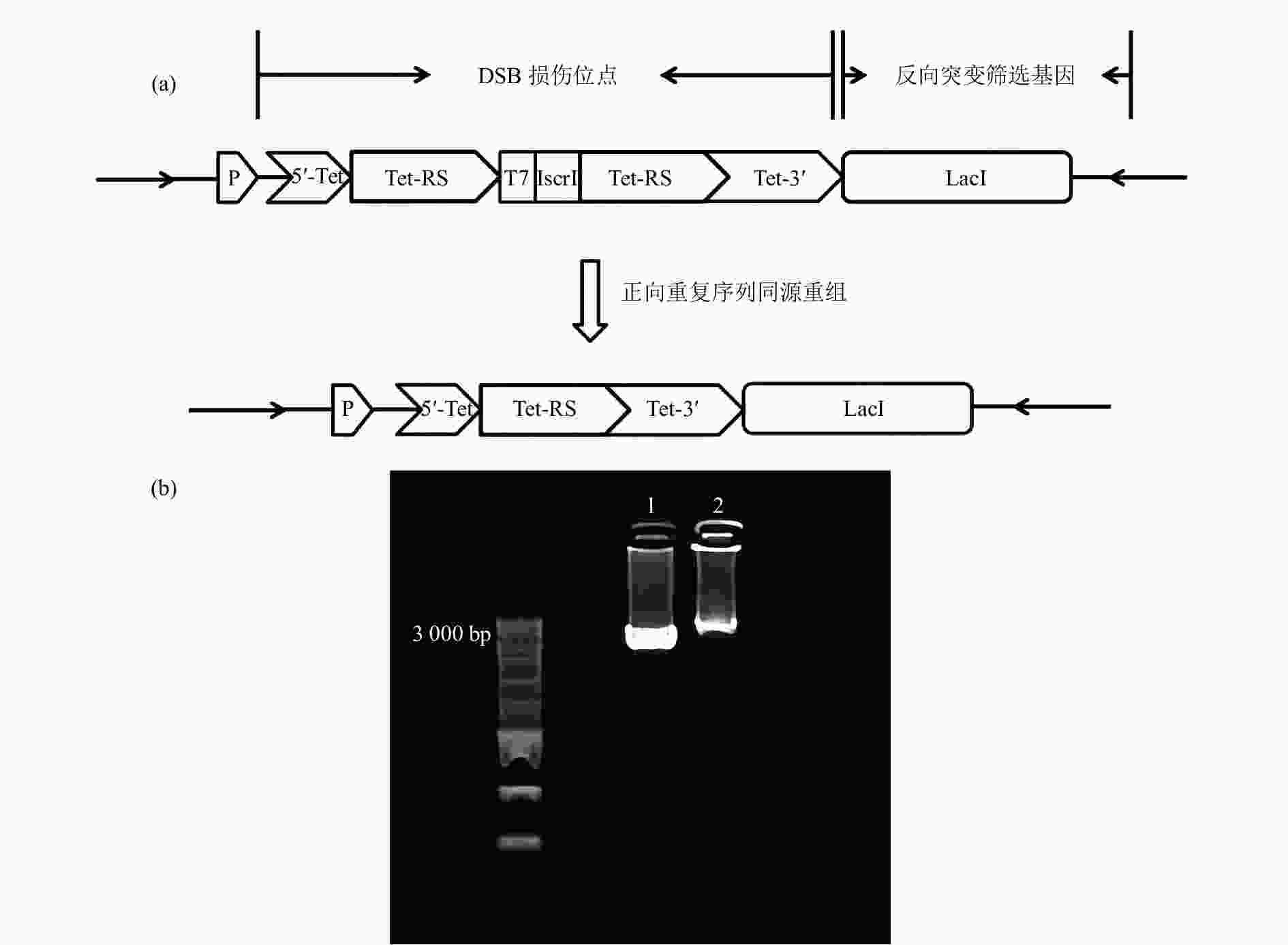
图 1 基于重复序列同源重组的反向突变筛选检测体系(TetA-LacI)的构建
接下来使用克隆载体PEASY-Blunt Cloning Kit的通用引物M13F&M13R对插入序列进行序列验证,首先挑选在LB+kan板的单克隆与LB+Kan+Tet筛选板上的单克隆进行PCR扩增并测序,检测构建的序列重组前后的准确性。电泳结果显示单条带,未发生同源重组的PCR产物片段长度应为3 411 bp,而发生同源重组的PCR产物片段长度应为2 811 bp[图1(b)]。测序结果显示重组前的序列构成和设计的序列一致,而在四环素抗性筛选板上的克隆测序结果显示TetA基因恢复成正确的序列,说明经四环素筛选到的单克隆为阳性克隆,该结果表明同源重组报告体系的可靠性。
-
同源重复片段长度影响同源重组的发生频率,分别构建了Tet-RS-300 bp、Tet-RS-600 bp和Tet-RS-900 bp不同同源重复片段长度的HRTetA报告基因。结果显示,随着HRTetA同源片段长度的增加,自发同源重组频率也在增加,表明片段越长,自发DSB的概率增加,同源重组频率相应增加[图2(a)]。HRTetA同源重复序列为900 bp时重组频率较高,可能原因在于TetA基因全长为1 200 bp,而在构建同源重复序列体系时5’-Tet加上900 bp的片段已经将近1 000 bp,有可能部分恢复TetA基因的活性,导致其未发生同源重组的情况下便可以在抗性板上筛选得到,筛选的准确性显著降低。而HRTetA同源重复序列为300和600 bp时本底同源重组频率均较低,在本实验中均以同源重复片段600 bp的TetA重组报告基因为基础。
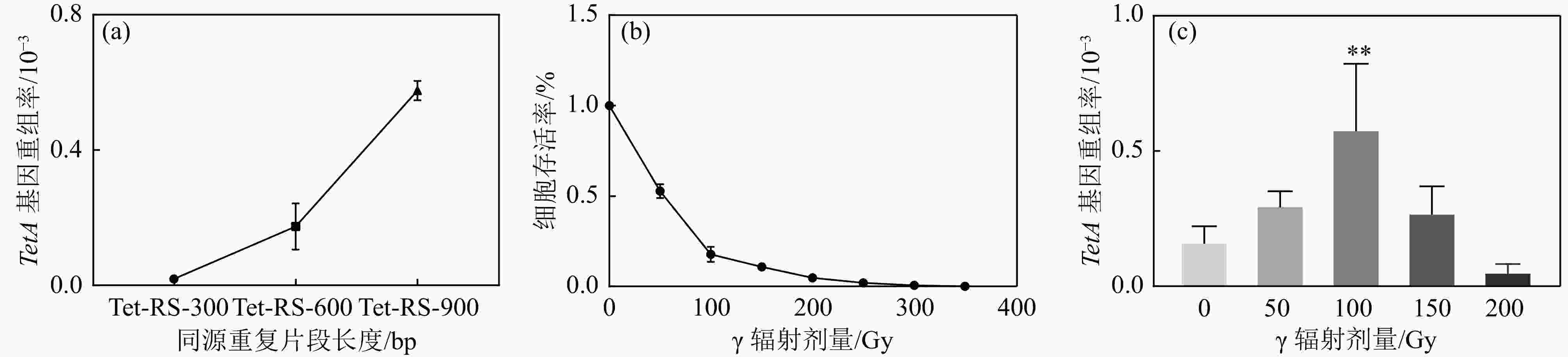
图 2 同源片段长度对辐射诱导同源重组频率的影响
接下来我们研究了含有同源重复片段长度为600 bp质粒的细胞辐射敏感性。结果显示,大肠杆菌细胞存活率随辐射剂量的增加而呈现下降的趋势[图2(b)]。进一步检测不同
$ \gamma $ 辐射剂量下的同源重组频率,结果发现,辐照前期HRTetA同源重复序列发生同源重组的频率和辐射剂量正相关,并在辐照剂量为100 Gy的时候呈现出极显著差异,而在150 Gy时急剧降低,这可能是由于辐照剂量过大对细胞引起的超杀效应引起的[图2(c)]。 -
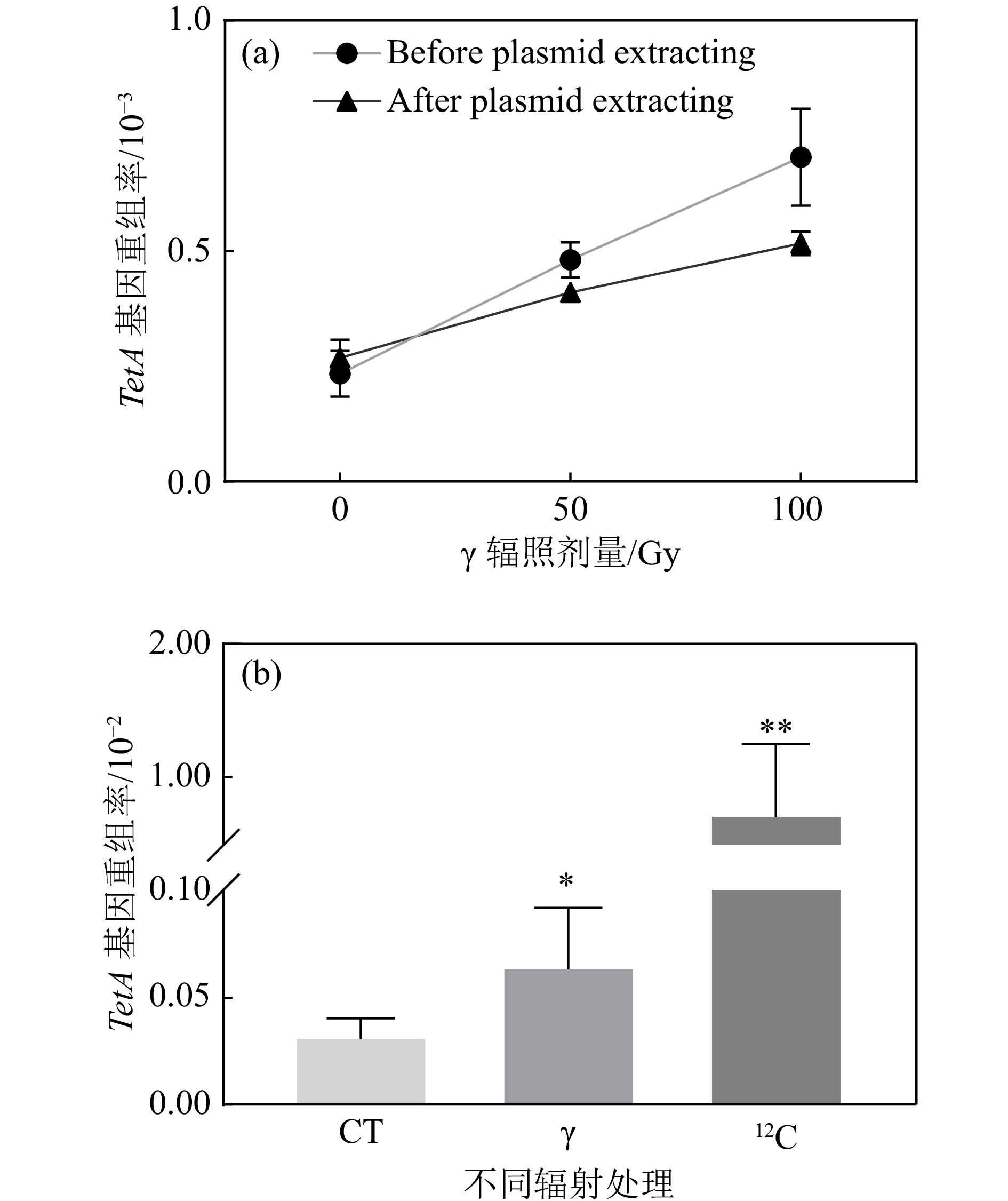
由于重组报告基因和反向突变筛选基因连接在质粒上,而细胞内的质粒是多拷贝的,即一个细胞中同时存在多个质粒,无法判断TetA基因发生同源重组和反向突变筛选基因LacI的突变是否发生在同一个质粒上。为了确保筛选的准确性,对辐照后的大肠杆菌细胞进行质粒提取并重新转化进入LacI缺失型大肠杆菌感受态细胞并进行TetA重组报告基因发生同源重组的筛选。经验证,质粒提取前后TetA重组率都呈现出剂量依赖性上升[图3(a)],表明质粒提取步骤不影响突变的分析。通过对
$ \gamma $ 与重离子辐照100 Gy后的TetA重组率比较发现,重离子辐射诱导TetA基因的同源重组频率远高于$ \gamma $ 辐射,相较于对照组以及$ \gamma $ 辐照100 Gy组,重离子辐照100 Gy 后诱导的TetA同源重组频率呈现量级的增加[图3(b)],表明重离子辐射诱导更多的DSB产生。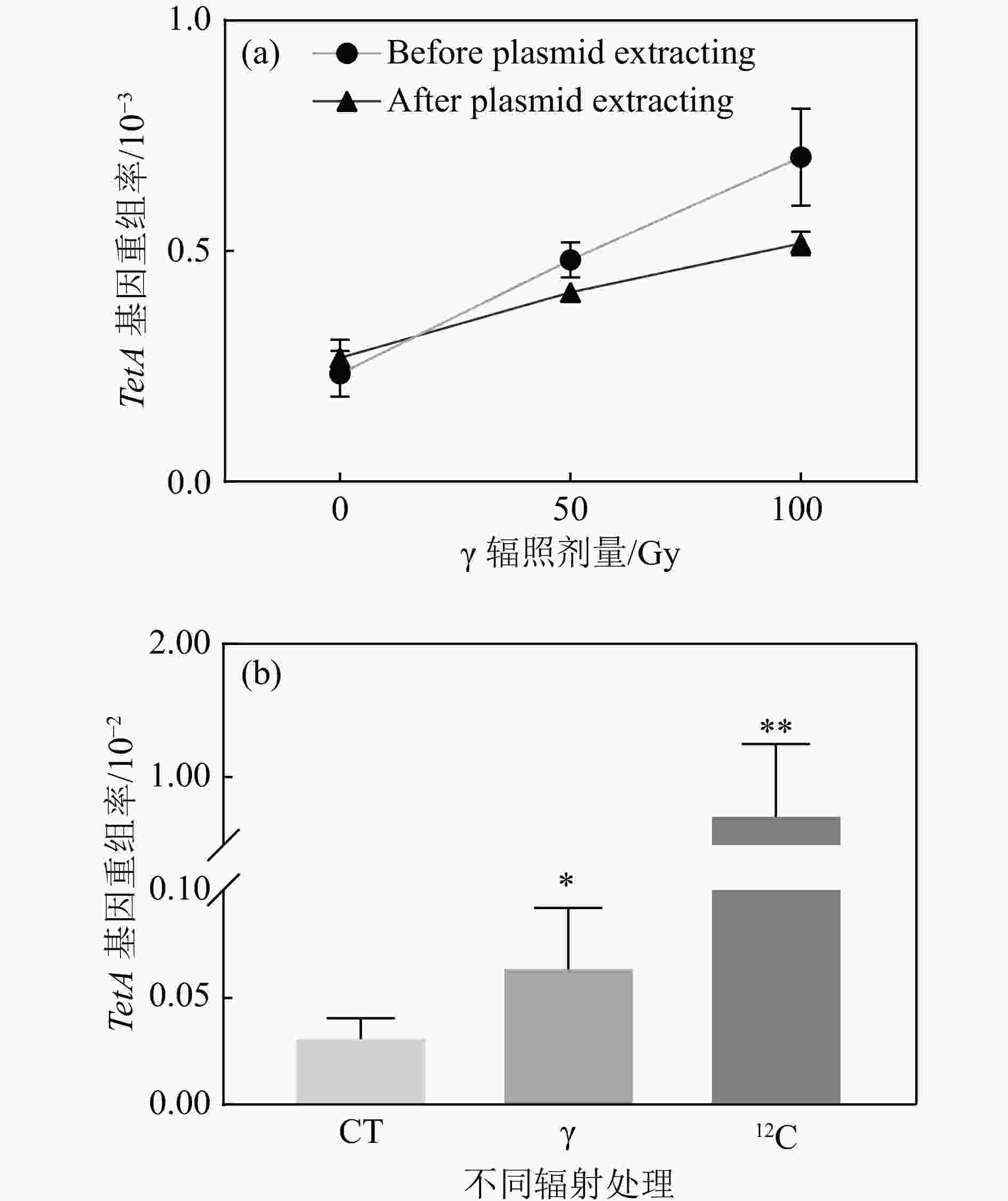
图 3 不同辐射剂量以及辐射类型对四环素同源重组频率的影响
-
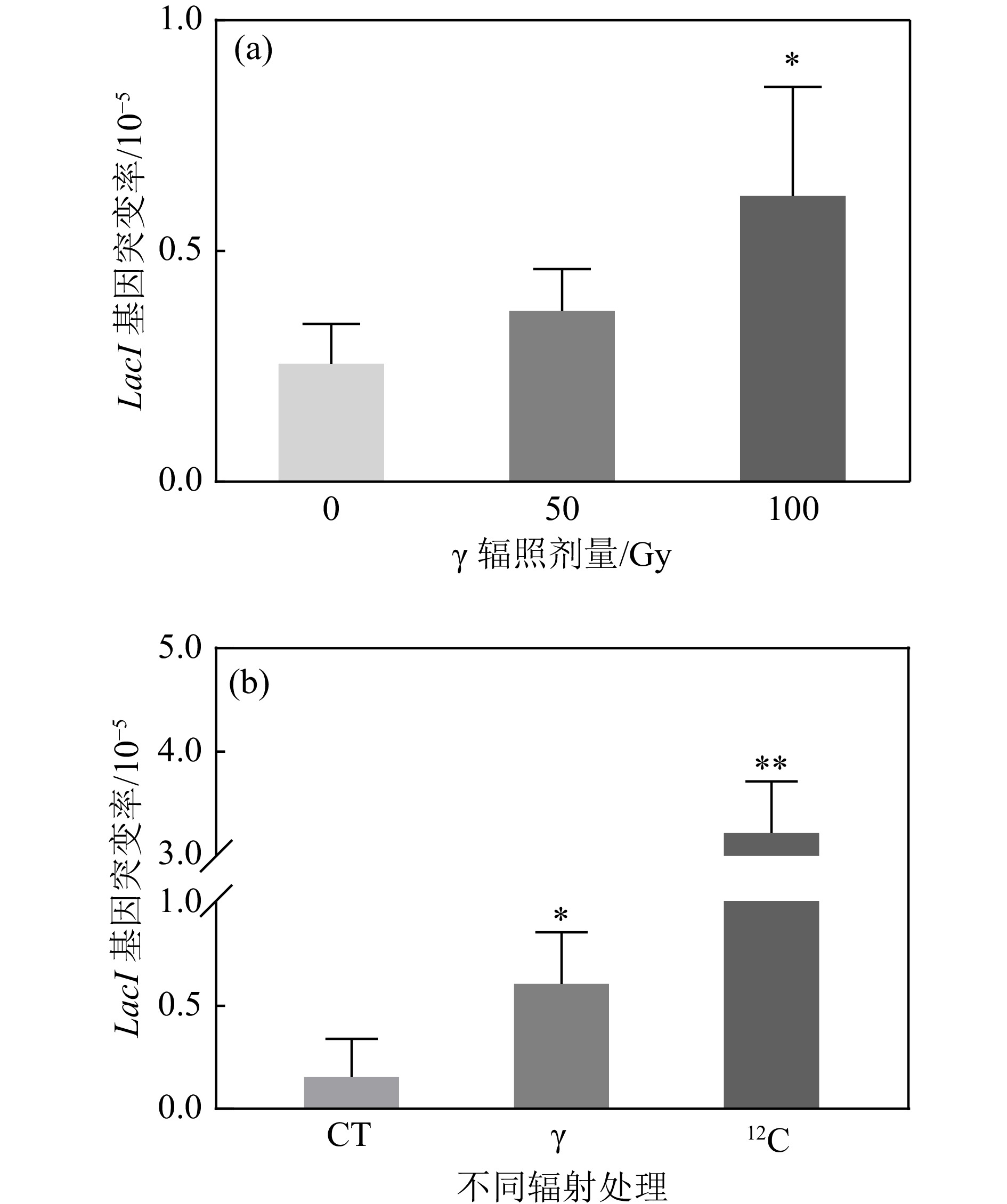
为了进一步对重离子辐射诱导团簇型DNA损伤进行分析,在TetA同源重组对DSB损伤位点进行定位以后,对其侧翼连接的反向突变筛选基因LacI进行序列分析,理论上如果损伤是离散的,则难以在DSB损伤位点检测到其侧翼序列的突变。LacI突变会抑制阻遏物的产生,通过营养缺陷型培养基P-Gal以及蓝白斑筛选的方法可以筛选到反向筛选基因LacI突变的质粒。首先我们检测了LacI基因单独响应辐射的情况,
$ \gamma $ 辐照后,LacI基因的突变频率随着辐照剂量的增加而增加,并在100 Gy时呈现显著差异[图4(a)]。通过对比100 Gy$ \gamma $ 与重离子辐照后的LacI基因突变情况,结果显示,重离子辐射诱导反向筛选基因LacI突变的频率是$ \gamma $ 辐照的5.2倍,是对照组的18.5倍,均呈现出极显著差异[图4(b)]。表明反向筛选基因LacI能够很好地响应辐射,且重离子辐照诱导更高频率的突变产生。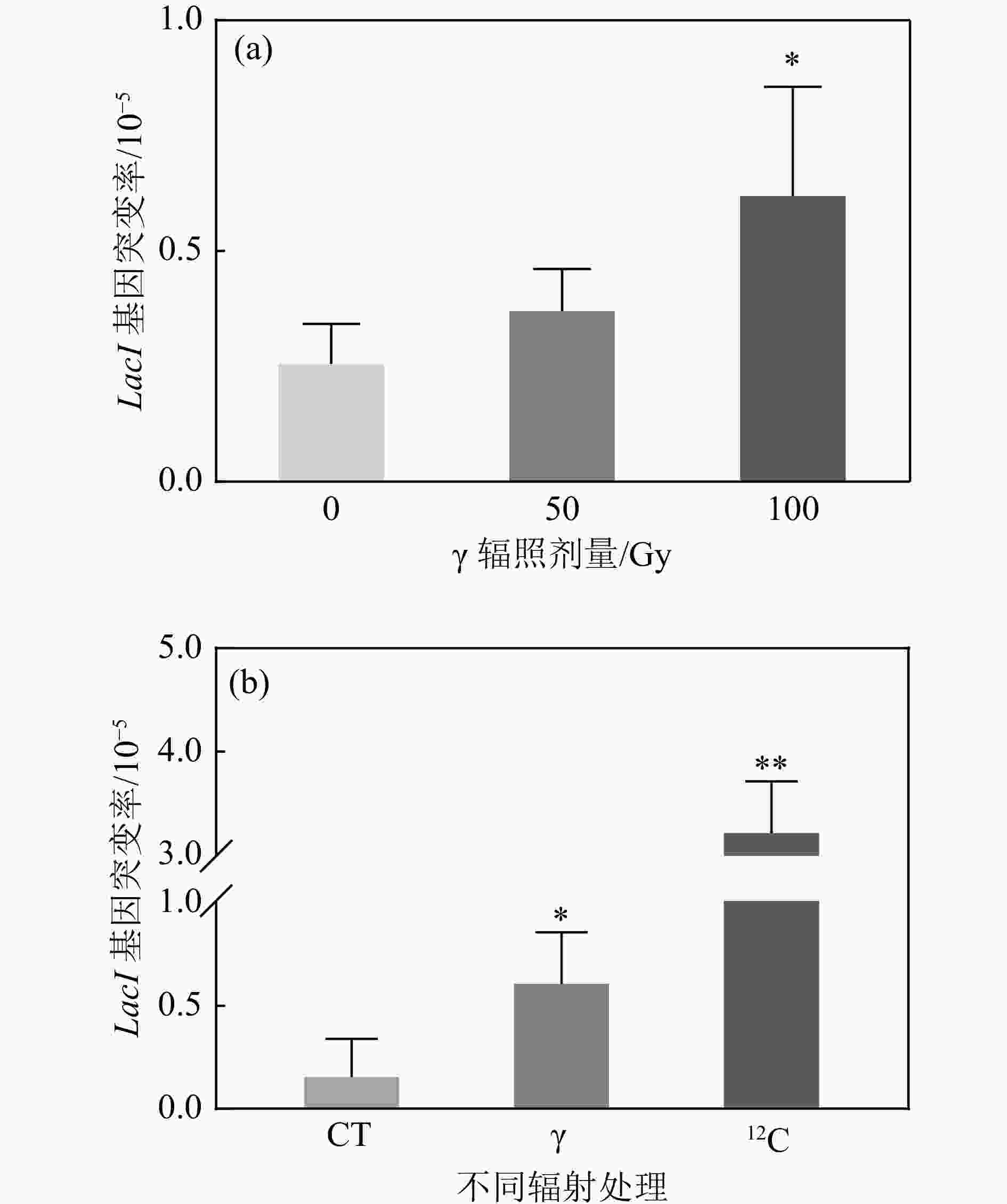
图 4 不同辐射剂量及辐射品质对LacI基因突变的影响
-
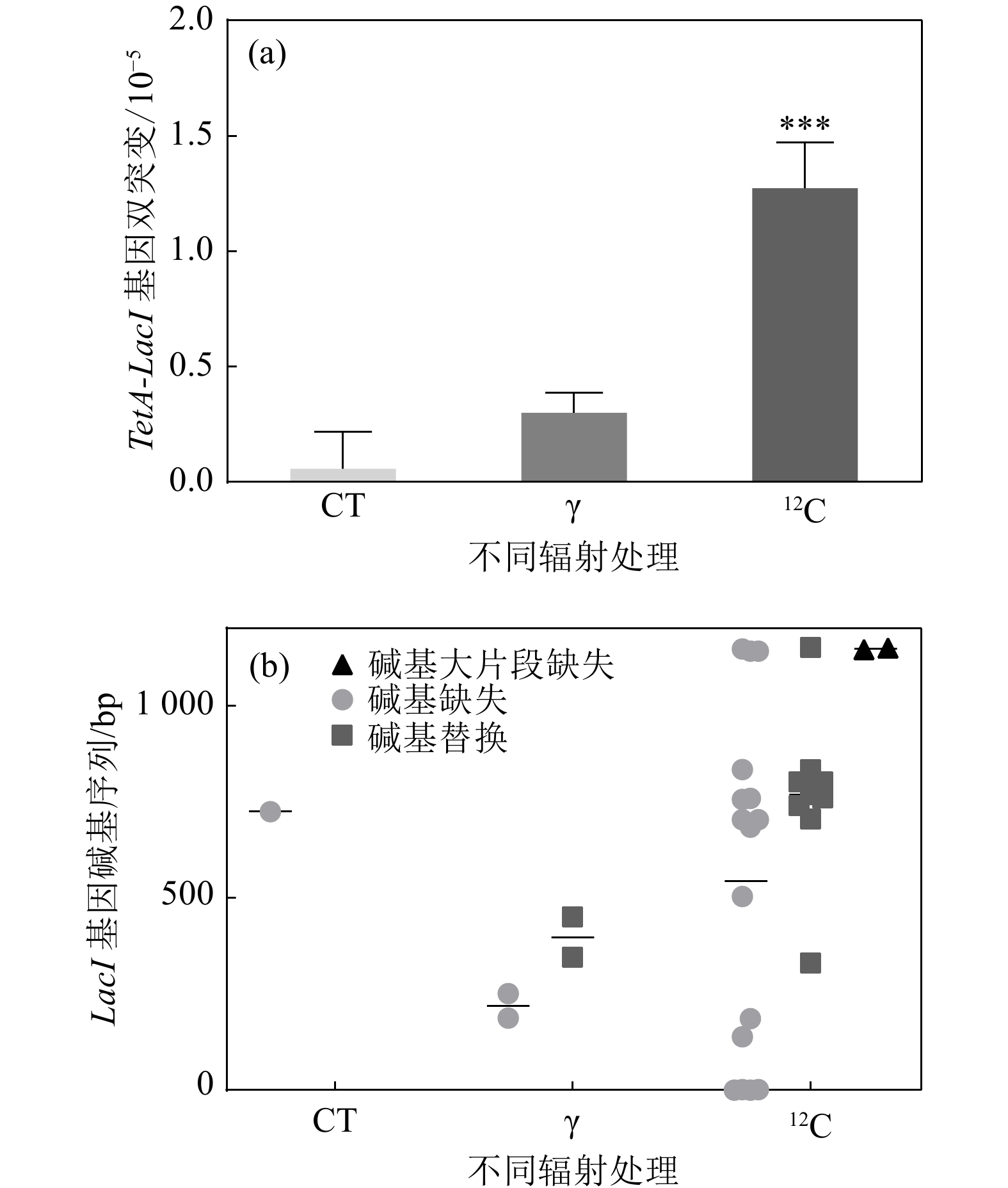
前期实验分别验证了四环素TetA基因辐照后发生同源重组的重组率,以及反向突变筛选基因LacI辐照后产生突变的突变率。接下来对
$ \gamma $ 辐照和重离子辐照后同时发生四环素TetA基因同源重组和反向突变筛选基因产生突变的情况进行分析。结果发现,重离子辐射诱导产生TetA基因重组且LacI基因突变的频率远高于对照组以及$ \gamma $ 辐照组,且重离子辐射诱导TetA基因以及LacI基因双突变产生是对照组的16倍,是$\gamma $ 辐射组的4倍,表明重离子辐射更高效地诱导团簇型损伤突变产生。将上述双筛突变的单克隆菌落进行LacI基因碱基序列测序,根据结果进一步分析重组报告基因侧翼序列的突变谱,如图5所示,重离子辐射诱导LacI基因产生的突变类型中,碱基缺失型突变比例较高,其次是碱基替换。在筛选到的突变体中,碱基缺失主要以A-为主,占碱基缺失突变的50%,而碱基替换中又以C-T之间的替换为主。并且相较于
$ \gamma $ 射线诱导产生的LacI基因突变,重离子辐射产生的突变位点较为集中。LacI基因序列长1 160 bp(包含启动子),筛选到的双突变克隆中,TetA基因发生同源重组的情况下侧翼序列LacI基因突变位点多集中在基因序列的650 bp之后,碱基缺失中的A-突变多集中在703 bp,而G-则集中在LacI基因的末端序列,从LacI基因全序列分析,碱基缺失中A-,G-热点位点周围多为连续的几个A碱基,推测与A-T之间的两个氢键相较于G-C之间的三个氢键更易断裂有关。相较于对照组以及$ \gamma $ 辐射组产生的LacI基因全部为单碱基突变,重离子辐射存在大片段缺失,并且位于LacI基因序列的中下游位置。以上结果表明,基于同源重组的反向突变基因筛选体系对于重离子辐射DNA损伤位点标记及突变分析实验方法是可行的。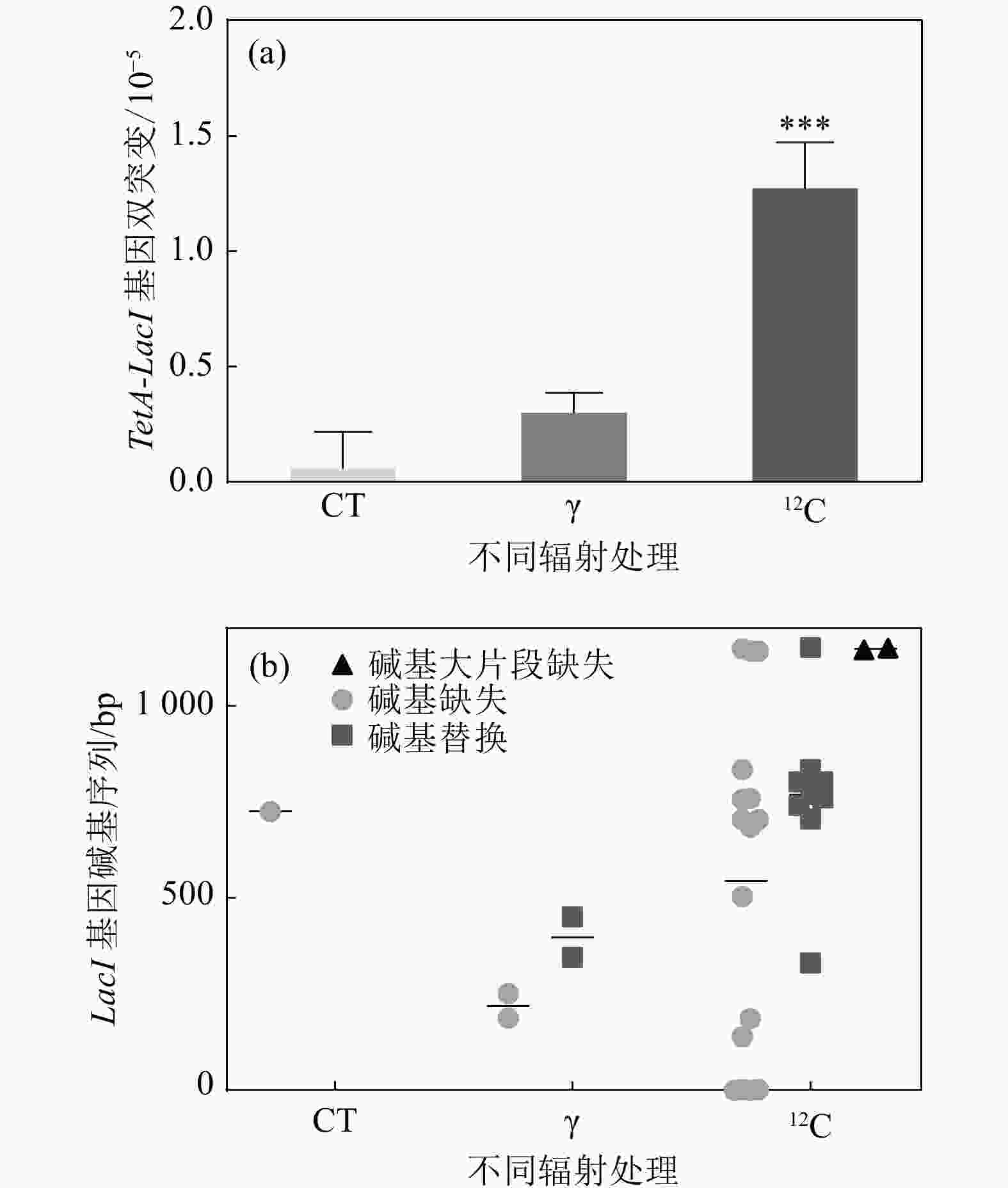
图 5 辐射品质对基于四环素同源重组的反向筛选体系的可行性的验证
-
重离子辐射的诱变机制一直是辐射生物学领域关注的焦点。重离子辐射具有很高的线传能密度和较宽的径向能量分布(Radial energy distribution),因此在击中DNA的靶点区域会产生团簇状的损伤。由于团簇损伤的构成比较多样,也就导致了团簇损伤修复具有更高的复杂性以及更高的致突变能力。研究重离子诱变机制的一个关键是能够在基因组序列水平确定重离子的辐射靶点。日本研究人员Hase等[16]和Kazama等[17]构建了叶毛调控基因突变体系和黄色荧光蛋白(YFP)植物体系对重离子辐射靶点进行定位,但是这两个体系存在一定的不足之处:一是实验过程繁琐,例如叶毛调控基因突变体系需要预先构建glp-1基因的杂合植株,YFP体系需要进行大量的后代筛选;二是体系使用的报告基因都是隐性突变,可以由多种损伤类型诱导,对重离子靶点序列的定位是非特异性的。基于HR修复对DSB特异性响应的特性,并且正向同源重复序列发生同源重组时是同源片段缺失型修复,在本研究中构建了TetA同源重组元件,同源序列任何位点发生DSB都可以导致重组的发生,具有很高的诱变效率,重组后的序列可以利用PCR片段长度进行区分[18]。因为重离子辐射诱导的团簇损伤的中心区域富集DSBs,因此同源重组报告基因主要用于重离子辐射靶点的序列定位。双突变筛选报告系是研究突变热点的重要方法,并在大肠杆菌、酵母等体系中得到应用,例如在酵母中,利用甲基甲磺酸盐诱导CAN1基因损伤,通过在CAN1基因两侧插入两个突变报告基因,确定DNA损伤诱发的突变范围以及检测多重突变发生的频率[19-21]。重离子诱发的DNA团簇损伤通常会导致聚集性的突变,这也是重离子辐射诱发突变的另一个重要特征[22]。基于此,本研究在重组报告基因的侧翼链接了LacI报告基因,进一步提高聚集性突变的检测效率。
在本研究中,低LET
$ \gamma $ 辐射处理显示TetA报告基因的同源重组率会随着辐照剂量的增加而上升,辐照剂量为100 Gy时产生显著性差异。而高LET重离子辐照产生的同源重组频率相较于$ \gamma $ 辐照有一个量级的增加,说明重离子辐照过程中产生更丰富的DSBs。对比重离子辐照与$ \gamma $ 辐照下侧翼LacI报告基因的突变频率,结果显示,重离子辐照下LacI基因的突变频率相较于$ \gamma $ 辐照同样也有一个量级的增加,显示重离子诱导的团簇损伤的周围也含有更多非DSB类型的损伤(也不能排除这种突变是DSBs修复所致)。在对实验体系进行双筛的情况下,即TetA基因同源重组和LacI基因突变同时发生,重离子辐照的结果也是远高于$ \gamma $ 辐照,双突变在重离子辐射诱导下出现的频率相较于$ \gamma $ 辐射有数倍的提升,而相较于对照组更是有一个量级的增加,这说明重离子辐射更倾向诱导DNA团簇型损伤。以上结果表明,基于同源重组的反向筛选体系能有效地对重离子辐射的靶点进行序列定位,并进一步分析DNA团簇损伤产生的突变模式。值得注意的是,$ \gamma $ 辐射也能诱导双突变的产生(尽管频率较低),说明该实验体系对重离子辐射的检测并不是完全特异性的,后期还需要结合LacI的突变的特征和模式做进一步的判断。另外,由于重离子辐射实验机时为一年两次,本研究在重离子辐射方面的数据还不够丰富,后期需要进一步结合重离子以及其它高LET辐射如质子等的物理特性来研究团簇损伤对径迹结构的依赖关系,以及通过构建不同间距的报告基因的实验体系来进一步研究团簇损伤导致突变的空间模式。总而言之,本研究提出了一种重离子辐射靶点定位和突变分析的研究思路,并基于大肠杆菌构建了相应的实验品系,通过
$\gamma $ 辐射和重离子辐射处理实验,我们证明了该研究思路的可行性,实验体系的建立也为进一步深入研究重离子辐射诱变的相关机制提供了方法学基础。
Construction and Validation of Experimental System for Heavy ion Radiation Targeted Loci Localization and lacI Mutation Analysis Based on Escherichia Coli
-
摘要: 重离子辐射具有独特的物理和生物学特性,在诱变育种等领域有着广泛的应用,但其诱变的机制并不完全清楚。不同于传统的 X 和
$ \gamma $ 射线,重离子辐射具有较高的线传能密度(Linear energy transfer, LET),主要诱导团簇状的DNA损伤,其演化为遗传变异的过程更为复杂,突变类型也更难预测。目前的实验技术很难在序列水平对重离子击中DNA的靶点进行定位,这致使重离子辐射诱变机制的研究相对滞后。针对这一问题,根据重离子辐射诱导的团簇损伤核心区域富含DNA双链断裂(Double-strand break, DSB)以及同源重组机制对DSB特异性响应的特性,首先构建了四环素抗性基因(TetA)同源重组元件用于确定DNA团簇损伤的序列定位,并在重组原件侧翼连接反向突变筛选基因LacI用于团簇损伤—突变的检测,最后把该质粒转化到大肠杆菌E.coli。在此基础上,比较分析$ \gamma $ 射线与碳重离子(80 MeV/u)辐照后同源重组和报告基因突变的情况,验证了该体系用于重离子辐射靶点序列定位及突变检测的可行性,为进一步研究重离子辐射诱变的相关机制奠定了方法学基础。-
关键词:
- 重离子 /
- $ \gamma $射线 /
- 同源重组 /
- 团簇损伤
Abstract: Heavy ion irradiation has unique physical and biological characteristics and has been widely applied in crop and bacteria mutation breeding. However, the mechanism underlying heavy ion irradiation mutagenesis is not completely clear. Unlike the conventional X and$ \gamma $ ray, heavy ion irradiation has a high linear energy transfer(LET) and mainly induces clustered DNA damage, the evolution of which to genetic variation is more complex and mutation types are more difficult to predict. Due to the limitation of experimental techniques, it is difficult to localize the DNA target of heavy ions at the level of DNA sequence. To address this issue, we first constructed a homologous recombination(HR) element of tetracycline resistance gene (TetA) in light of high abundance of DNA double strand breaks(DSB) in the core of heavy ion radiation-induced cluster damage. And the DSB specific response of HR. We then linked a reverse mutation screening gene lacI to the TetA recombination elements to detect cluster damage-derived mutations. Finally, this plasmid was transformed into E.coli. Based on this experimental system we comparatively analyzed TetA recombinations and mutations of LacI gene after irradiation with$ \gamma $ -ray and carbon heavy ions (80 MeV/u). We preliminarily verified the feasibility of this research strategy and provided a methodological foundation for further investigation of the mechanisms underlying heavy ion radiation mutagenesis.-
Key words:
- heavy ions /
- $ \gamma $-ray /
- homologous recombination /
- clustered damage
-
图 1 基于重复序列同源重组的反向突变筛选检测体系(TetA-LacI)的构建
(a) TetA-LacI体系构建原理图,以Tet-RS为600 bp为例;(b) TetA-LacI体系序列验证。条带1为同源重组后的片段;条带2为同源重组前的片段。
图 2 同源片段长度对辐射诱导同源重组频率的影响
(a) TetA同源序列长度的自发重组率比较;(b) 细胞存活率随辐照剂量的增加而降低;(c) 不同剂量$ \gamma $辐射对TetA基因同源重组率的影响;**P<0.01。
图 3 不同辐射剂量以及辐射类型对四环素同源重组频率的影响
(a) $ \gamma $辐照后提质粒转化对四环素重组率的影响;(b)不同辐射类型辐照100 Gy处理后TetA同源重组率的比较。**P<0.01,*P<0.05。
图 4 不同辐射剂量及辐射品质对LacI基因突变的影响
(a) 不同剂量$ \gamma $辐照后LacI基因突变率;(b) 不同辐射类型辐照100 Gy处理对LacI基因突变率的影响;**P<0.01,*P<0.05。
图 5 辐射品质对基于四环素同源重组的反向筛选体系的可行性的验证
(a) 不同辐射处理诱导同源重组双靶点体系突变率;(b) LacI基因序列上突变点定位及突变类型;***P<0.001。
表 1 融合PCR引物列表
引物名称 引物序列 TetA-2F GCTTGACACTTTATCACTGATAAAC TetA-R TCAGCGATCGGCTCGTTG LacI-F ACACCATCGAATGGCGCAAAACC LacI-R TCACTGCCCGCTTTCCAGTCGG LacIF-TetR-R GGTTTTGCGCCATTCGATGGTGTTCAGCGATCGGCTCGTTG TetR-LacIF-F CAACGAGCCGATCGCTGAACACCATCGAATGGCGCAAAACC  下载: 导出CSV
下载: 导出CSV
-
[1] JIN Yuanchang, LI Jingwen, LI Jieyun, et al. Front Oncol, 2021, 11: 634913. doi: 10.3389/fonc.2021.634913 [2] NICKOLOFF J, SHARMA N, TAYLOR L. Genes (Basel), 2020, 11(1): 99. doi: 10.3390/genes11010099 [3] IFIGENEIA V, ZACHARENIA N, ALEXANDROS G, et al. Cancers (Basel), 2019, 11(11): 1789. doi: 10.3390/cancers11111789 [4] RYUICHI O. International Journal of Cancer, 2012, 130(5): 991. doi: 10.1002/ijc.26445 [5] JULIE A, NIDHI B, CRISTINA M, et al. Antioxid Redox Signal, 2014, 21(2): 260. doi: 10.1089/ars.2013.5489 [6] LI Yanqiu, KARDELL M B, WANG Feifei, et al. DNA Repair (Amst), 2021, 108: 103244. doi: 10.1016/j.dnarep.2021.103244 [7] STEINERT J, SCHIML S, PUCHTA H. Plant Cell Rep, 2016, 35(7): 1429. doi: 10.1007/s00299-016-1981-3 [8] ALEJANDRA B. PLoS One, 2021, 16(4): e0237413. doi: 10.1371/journal.pone.0237413 [9] BOYKO A, ZEMP F, FILKOWSKI J, et al. Plant Physiol, 2006, 141: 488. doi: 10.1104/pp.105.074658 [10] BOYKO A, KOVALCHUK I. Curr Opin Plant Boil, 2011, 14: 260. doi: 10.1016/j.pbi.2011.03.003.doi:10.1016/j.pbi.2011.03.003 [11] KOVALCHUK I, KOVALCHUK O, ARKHIPOV A. Nat Biotechnol, 1998, 16: 1054. doi: 10.1038/3505 [12] KOVALCHUK O, KOVALCHUK I, TITOV V. Mutat Res, 1999, 446: 49. doi: 10.1016/s1383-5718(99)00147-3 [13] KOVALCHUK I, KOVALCHUK O, HOHN B. Trends in Plant Sci, 2001, 6: 306. doi: 10.1016/S1360-1385(01)01985-9 [14] VIKASH K, CORENTIN C. Front Cell Dev Biol, 2021, 9: 642737. doi: 10.3389/fcell.2021.642737 [15] JOSEPH S, RUSSELL D. CSH Protoc, 2006, 2006(1): 3944. doi: 10.1101/pdb.prot3944 [16] HASE Y, YOSHIHARA R, NOZAWA S, et al. Mutat Res, 2012, 731: 41. doi: 10.1016/j.mrfmmm.2011.10.004 [17] KAZAMA Y, SAITO H, FUJIWARA M, et al. Biosci Biotechnol Biochem, 2007, 71: 2864. doi: 10.1271/bbb.70571 [18] WRIGHT W, SHAH S, HEYER W. J Biol Chem, 2018, 293(27): 10524. doi: 10.1074/jbc.TM118.000372 [19] YANG Y, GORDENIN D, RESNICK M. DNA Repair, 2010, 9(8): 914. doi: 10.1016/j.dnarep.2010.06.005 [20] MOORE J M, CORREA R, ROSENBERG S M, et al. PLoS Genet, 2017, 13(7): e1006733. doi: 10.1371/journal.pgen.1000264 [21] YANG Y, STERLING J, STORICI F, et al. PLoS Genet, 2008, 4(11): e1000264. doi: 10.1073/pnas.1104681108 [22] ASAITHAMBY A, CHEN D. Mutat Res, 2011, 711(1-2): 87. doi: 10.1016/j.mrfmmm.2010.11.002 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 411
- HTML全文浏览量: 306
- PDF下载量: 30
- 被引次数: 0



























 甘公网安备 62010202000723号
甘公网安备 62010202000723号